6.6 富集分析 EMP_enrich_analysis
富集分析是一种生物信息学方法,用于确定一组基因或蛋白质在特定的生物学通路或功能中是否显著富集。数据库包含了大量的基因和基因产物的功能信息,以及这些基因在代谢通路、信号传导通路和疾病通路中的作用。本模块可以在线读取KEGG、Go、Reactome等数据库信息,完成富集分析的常规步骤。
6.6.1 基于KO/EC注释的KEGG富集分析
宏基因组微生物数据分析过程中可以得到KO/EC的功能基因注释结果,使用模块EMP_diff_analysis可获取差异基因,并利用模块EMP_enrich_analysis进行KEGG富集分析。
注意:
①参数
②参数
③参数
①参数
KEGG_Type可以指定按照pathway富集(KEGG_Type = 'KEGG')或者Module富集(KEGG_Type = 'MKEGG')。②参数
species默认使用全物种数据作为背景基因(species = 'all'),也可以指定具体物种数据作为背景基因进行富集。③参数
condition根据pvalue或者校正p值筛选差异基因,并进行富集。
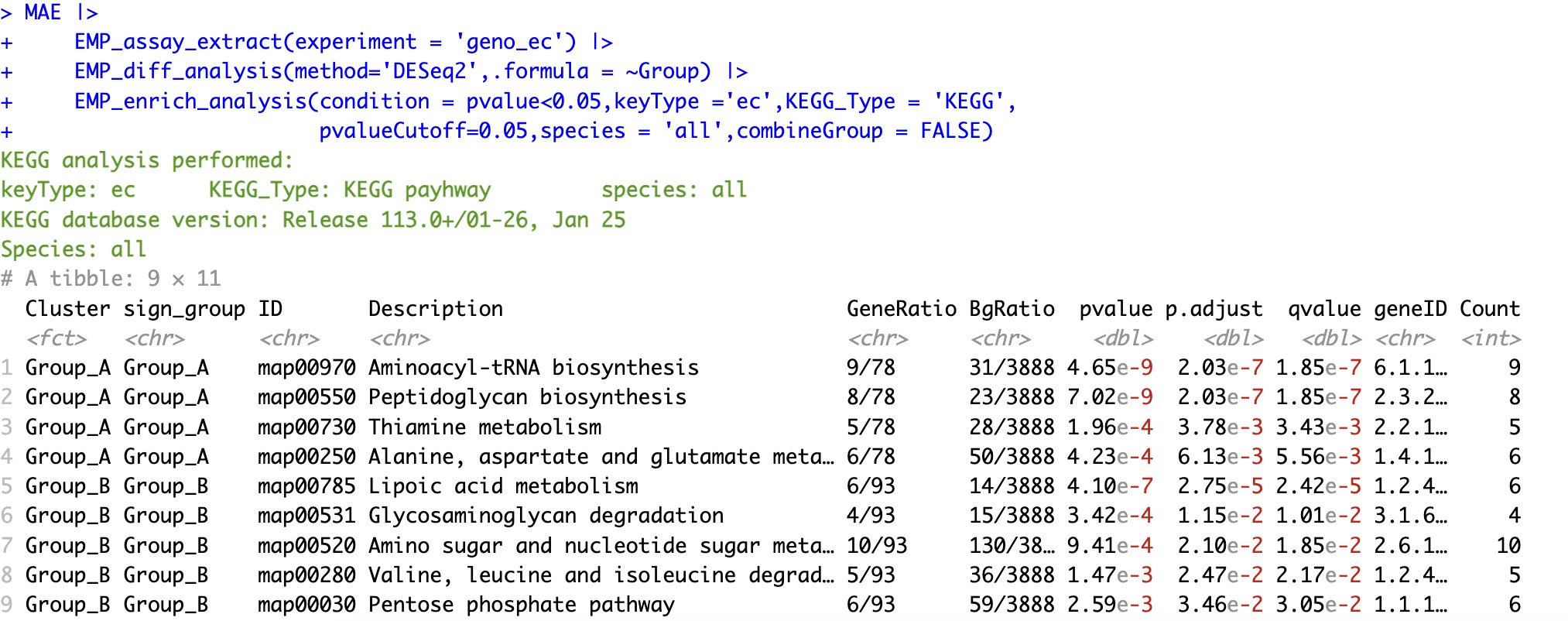
🏷️示例1:
MAE |>
EMP_assay_extract(experiment = 'geno_ec') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(condition = pvalue<0.05,keyType ='ec',KEGG_Type = 'KEGG',
pvalueCutoff=0.05,species = 'all',combineGroup = FALSE)
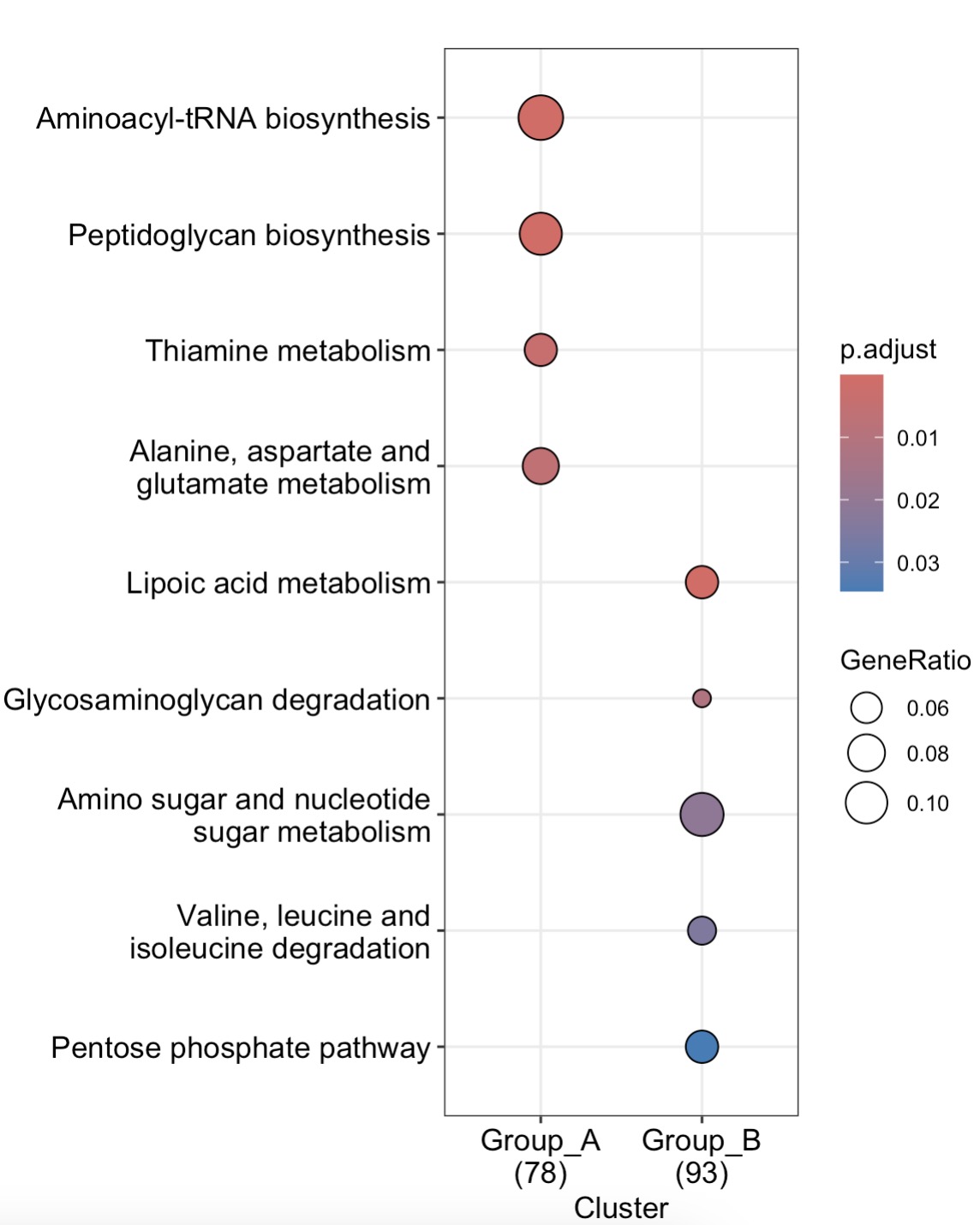
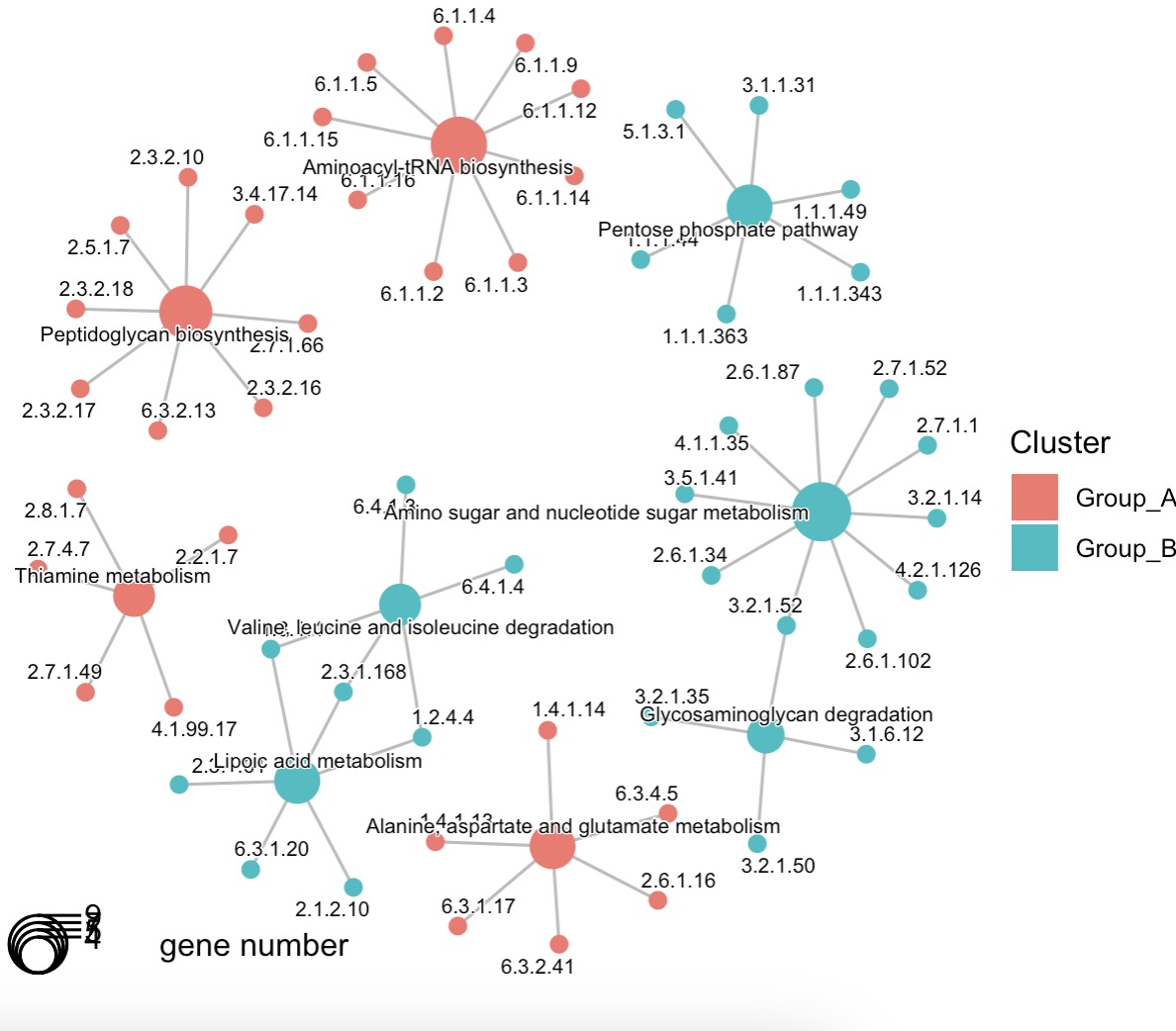
🏷️示例2:模块EMP_enrich_dotplot和EMP_enrich_netplot继承了enrichplot包的dotplot和cnetplot,可以对富集分析的结果进行可视化。
MAE |>
EMP_assay_extract(experiment = 'geno_ec') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(condition = pvalue<0.05,keyType ='ec',KEGG_Type = 'KEGG',
pvalueCutoff=0.05,species = 'all',combineGroup = FALSE) |>
EMP_enrich_dotplot()
MAE |>
EMP_assay_extract(experiment = 'geno_ec') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(condition = pvalue<0.05,keyType ='ec',KEGG_Type = 'KEGG',
pvalueCutoff=0.05,species = 'all',combineGroup = FALSE) |>
EMP_enrich_netplot()
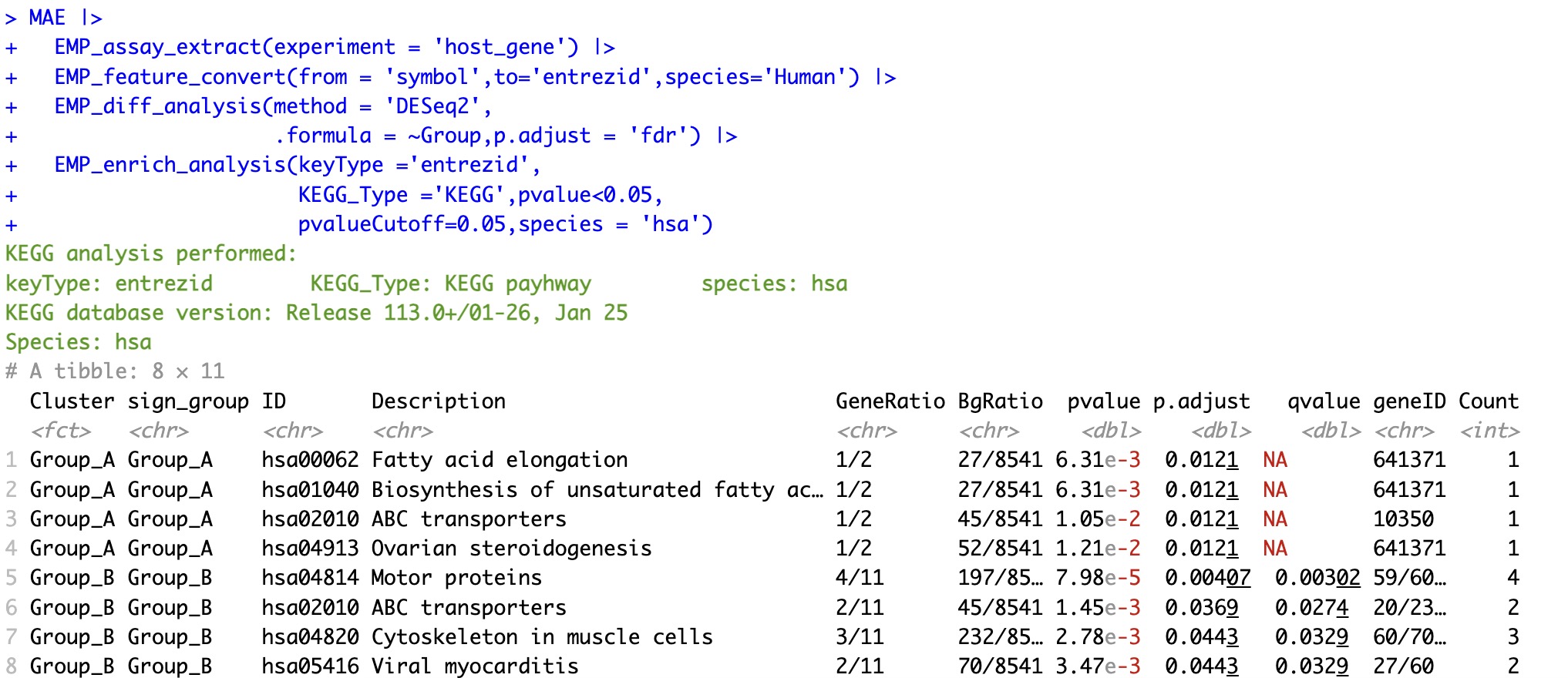
6.6.2 基于基因名称的的KEGG富集分析
这类数据常见于宿主组织的Bulk 转录组数据。在进行分析前,需要使用模块EMP_feature_convert将基因的symbol转换成entrezid。
🏷️示例:
MAE |>
EMP_assay_extract(experiment = 'host_gene') |>
EMP_feature_convert(from = 'symbol',to='entrezid',species='Human') |>
EMP_diff_analysis(method = 'DESeq2',
.formula = ~Group,p.adjust = 'fdr') |>
EMP_enrich_analysis(keyType ='entrezid',
KEGG_Type ='KEGG',pvalue<0.05,
pvalueCutoff=0.05,species = 'hsa')
6.6.3 基于代谢产物的KEGG富集分析
这类数据主要是代谢组学产生,富集分析前需要将特征名称转换为compund ID。
🏷️示例:
MAE |>
EMP_assay_extract(experiment = 'untarget_metabol')|>
EMP_collapse(na_string=c('NA','null','','-'),
estimate_group = 'MS2kegg',method = 'sum',collapse_by = 'row') |>
EMP_diff_analysis(method='DESeq2',.formula = ~Group) |>
EMP_enrich_analysis(keyType ='cpd',
KEGG_Type ='KEGG',pvalue<0.05,
pvalueCutoff=0.05,species = 'all')

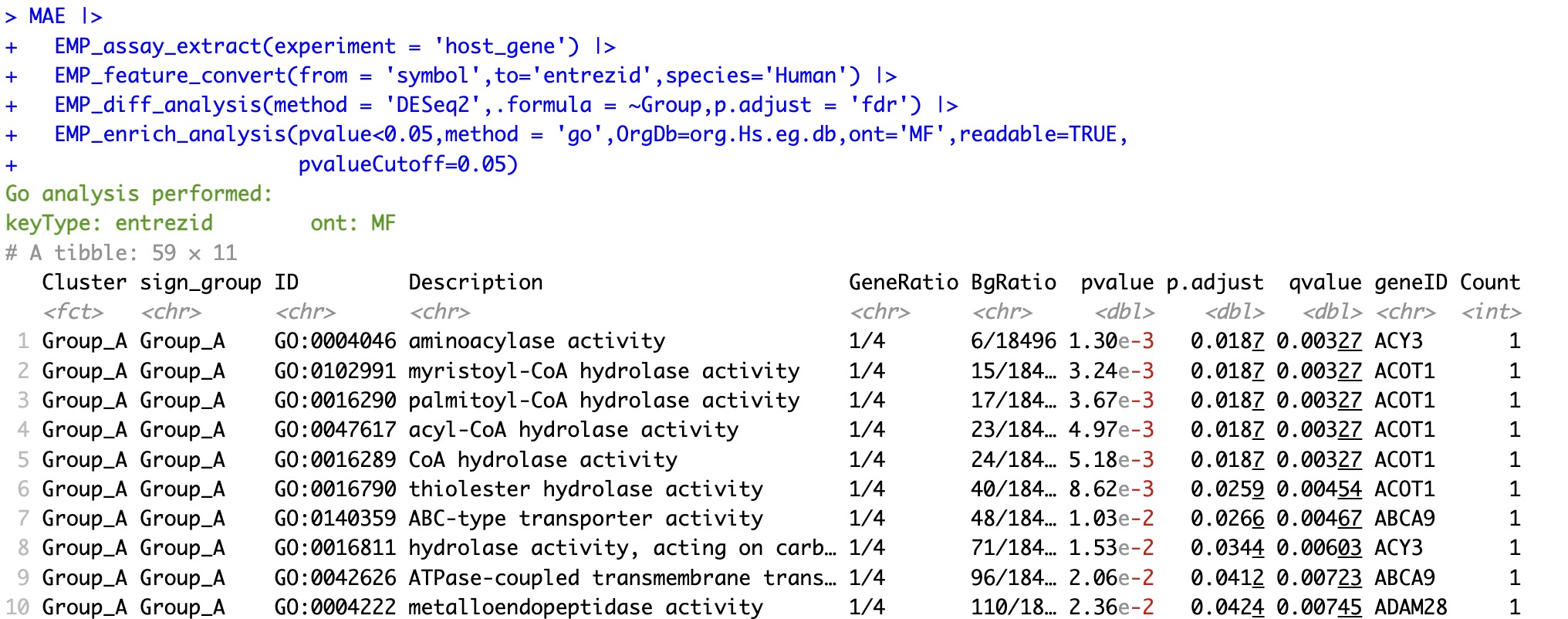
6.6.4 基于基因名称的的Go富集分析
注意:
①在Go分析中需要参数
②参数
③参数
①在Go分析中需要参数
OrgDb来指定物种。②参数
ont可以实现BP(生物过程),MF(分子功能),CC(细胞组分)和ALL的富集方式。③参数
readable可以将富集结果中的entrezid转换为symbol。
🏷️示例:
library(org.Hs.eg.db)
MAE |>
EMP_assay_extract(experiment = 'host_gene') |>
EMP_feature_convert(from = 'symbol',to='entrezid',species='Human') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group,p.adjust = 'fdr') |>
EMP_enrich_analysis(pvalue<0.05,method = 'go',OrgDb=org.Hs.eg.db,ont='MF',readable=TRUE,
pvalueCutoff=0.05)
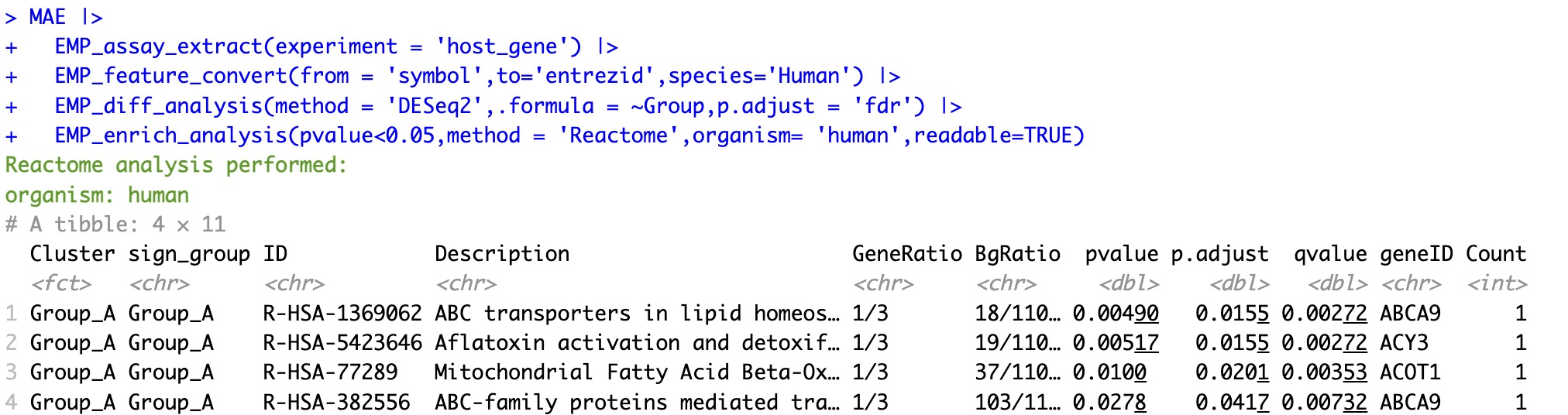
6.6.5 基于基因名称的的Reactome富集分析
注意:
①在Reactome分析中需要参数
②参数
①在Reactome分析中需要参数
organism来指定物种。②参数
readable可以将富集结果中的entrezid转换为symbol。
🏷️示例:
MAE |>
EMP_assay_extract(experiment = 'host_gene') |>
EMP_feature_convert(from = 'symbol',to='entrezid',species='Human') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group,p.adjust = 'fdr') |>
EMP_enrich_analysis(pvalue<0.05,method = 'Reactome',organism= 'human',readable=TRUE)
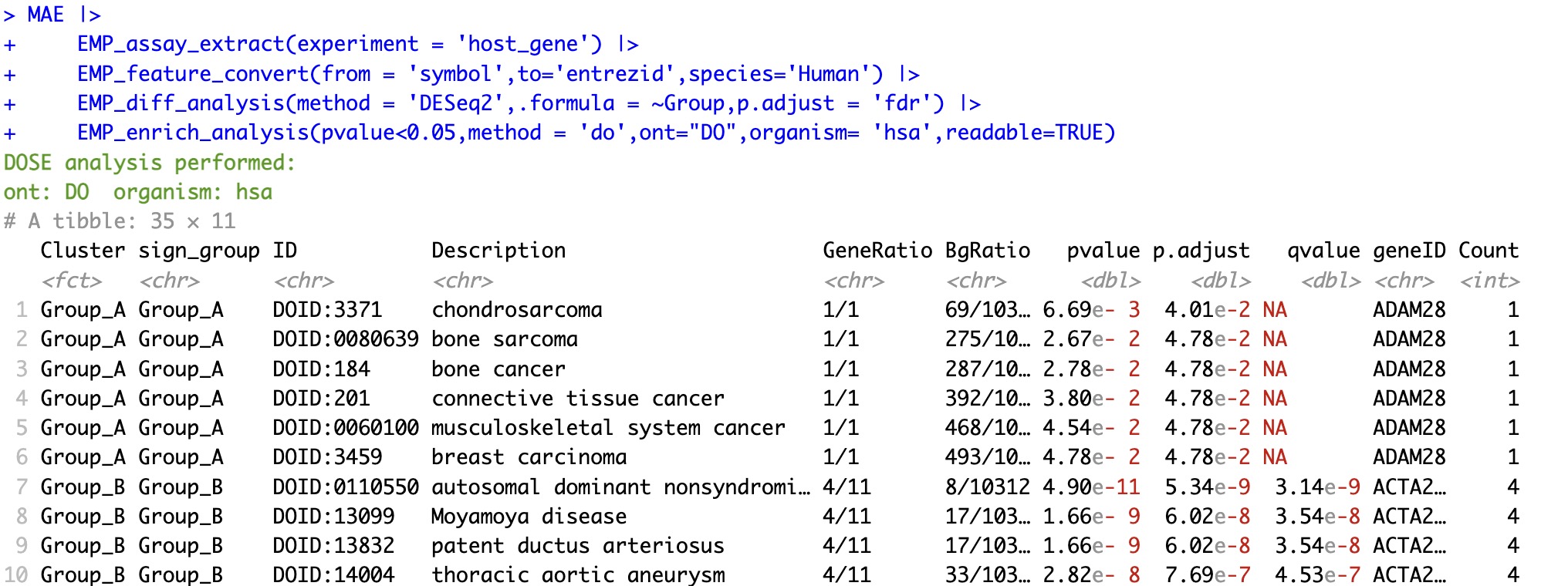
6.6.6 基于基因名称的的DOSE富集分析
注意:
①在DOSE分析中需要参数
②参数
①在DOSE分析中需要参数
organism来指定物种。注意此参数与Reactome的写法不同②参数
ont支持HDO,HPO和MPO模式。
🏷️示例:
MAE |>
EMP_assay_extract(experiment = 'host_gene') |>
EMP_feature_convert(from = 'symbol',to='entrezid',species='Human') |>
EMP_diff_analysis(method = 'DESeq2',.formula = ~Group,p.adjust = 'fdr') |>
EMP_enrich_analysis(pvalue<0.05,method = 'do',ont="HDO",organism= 'hsa',readable=TRUE)